好文推荐 | Aβ寡聚体与阿尔茨海默病研究进展
2023-10-24 中风与神经疾病杂志 中风与神经疾病杂志 发表于上海

本文综述了对Aβ寡聚体的新近认知,以及它们的结构特点和致病作用,以求更好的理解Aβ寡聚体和AD的关系,为之后的研究提供可能的方向。
摘要:阿尔茨海默病(Alzheimer's disease,AD)是一种以记忆损害为主要表现渐进性发展的神经退行性疾病,随着老龄化社会发展发病率呈上升趋势,带来巨大的社会经济负担。近年来的研究使AD经典发病机制淀粉样蛋白级联假说受到了挑战,Aβ寡聚体(Aβ oligomers,AβOs)的毒性作用得到了一致性的认可,可能是AD发病的触发因素。关于Aβ寡聚体具体来源、结构和致病机制的认识还不充分,可能不同类型寡聚体致病毒性不一致,作用机制有差别。本文综述了对Aβ寡聚体的新近认知,以及它们的结构特点和致病作用,以求更好的理解Aβ寡聚体和AD的关系,为之后的研究提供可能的方向。
阿尔茨海默病(Alzheimer's disease,AD)是最常见痴呆症类型,占痴呆症病例的近60%~80%,以早期显著的情景记忆损害为主要表现,并伴有学习记忆能力下降、精神行为异常、语言受损、执行功能受损等广泛的认知功能损害。关于AD的发病机制提出很多假说,其中淀粉样蛋白级联假说认为β淀粉样蛋白(amyloid β-protein,Aβ)的过量产生并在脑内沉积是AD的发病中心环节,随着研究的进展发现淀粉样蛋白级联假说并不完善,比如正常老年人脑内存在老年斑(Senile plaque,SP),但是认知功能正常,针对Aβ的被动免疫治疗可以清除老年斑,但并不改善认知。这表明AD脑内特定区域的老年斑数量与局部神经元死亡、突触丢失程度、认知功能障碍相关性并不好,近来研究表明可溶性Aβ寡聚体(Aβ oligomers,AβOs)水平与突触丢失、认知功能障碍严重程度之间有更好的相关性,Aβ在脑内聚集形成Aβ寡聚体的毒性作用得到了一致的认可,并且出现了大量的关于其来源、结构、作用机制和应用前景的研究。
1 Aβ与SP、tau
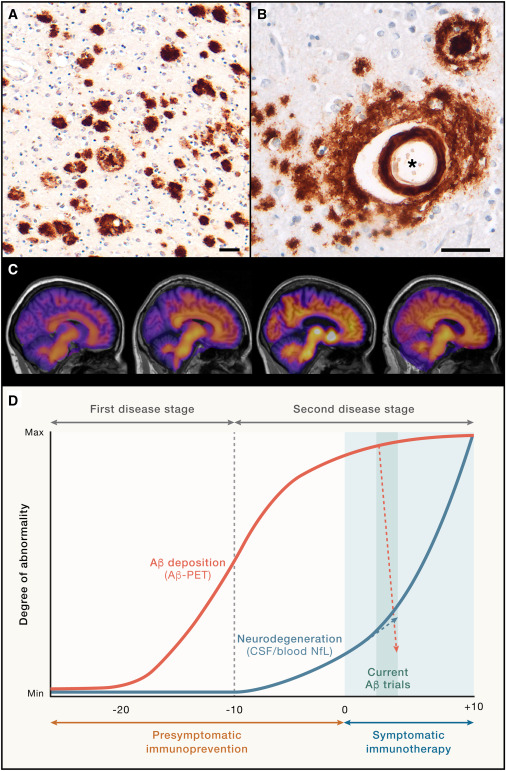
Aβ单体易于聚集,最初认为Aβ寡聚体是单体向纤维体聚集的中间类型,直到实验发现Aβ寡聚体可以独立于纤维体稳定存在,并表现出强烈的神经毒性。Aβ寡聚体在人脑和动物模型中广泛存在,并且其形成要早于老年斑,这一点已经被多种实验方法所证实。随之“Aβ寡聚体假说”被广泛接受,其认为Aβ寡聚体是AD脑内最重要的神经毒性物质,最终导致了突触的损伤和认知障碍。SP是AD的组织病理学标志,可溶性Aβ寡聚体可以由AD脑中的SP向周围扩散。小鼠模型显示可溶性寡聚体在密集老年斑附近浓度较高,随着距离斑块越远,Aβ寡聚体浓度下降。随着AD进展,单个斑块体积最终保持稳定而不再增加,表明致密斑块中不溶性Aβ可以转变为可溶形式的Aβ并释放到周围脑中,这些观点支持了淀粉样蛋白沉积与可溶性Aβ寡聚体之间存在动态平衡的观点。Aβ寡聚体可能是导致痴呆发生和神经元损伤的触发因素,而tau蛋白磷酸化为导致AD相关突触损伤的直接原因,可溶性Aβ寡聚体是病理性tau蛋白磷酸化的上游激活因素,并最终导致神经元死亡。研究一致认为Aβ寡聚体通过酪氨酸蛋白激酶(Fyn)介导了tau蛋白异常磷酸化及神经原纤维缠结的形成。
2 Aβ寡聚体的毒性作用
Roher等于1996年首次报道了可溶性寡聚体可能与AD相关,Aβ寡聚体是强烈的毒性物质,甚至在纳摩尔浓度水平可以杀死海马切片中的成熟神经元。动物侧脑室注射Aβ寡聚体损伤海马突触长时程增强(Long-term Potentiation,LTP)和空间学习能力。研究表明突触可塑性和认知功能密切相关,尤其是在轻度认知功能障碍(MCI)阶段,同时注射Aβ寡聚体和抗Aβ抗体6E10会中和寡聚体诱导的LTP功能障碍。体外应用天然提取的寡聚体制剂可以导致树突棘快速丧失和突触可塑性缺陷,而颅内注射类似制剂会损害学习和记忆。Cheng等在体外试验研究中发现与野生型小鼠Aβ相比,Arctic突变(AβE22G)加速了Aβ纤维化,但降低了Aβ寡聚体的数量。在携带人淀粉状蛋白前体蛋白(hAPP)转基因小鼠中,加入Arctic突变显著增强Aβ斑块的形成但降低特定Aβ寡聚体的数量。在过度表达Arctic突变体和野生型小鼠中,当它们体内的Aβ寡聚体水平相一致时具有相似的行为异常和神经元缺陷,但是斑块负荷相差甚远。与纤维状Aβ沉积斑块相比,Aβ寡聚体是hAPP转基因小鼠功能缺陷更可能的决定因素,同时Aβ寡聚体的毒性作用在人体内也得到了证实。
近来以焦谷氨酸(AβpE3)开始的N-末端截短的Aβ肽受到了关注,其代表了AD大脑内Aβ的主要成分,与全肽段Aβ相比,AβpE3 较Aβ 1-42具有更高的聚集倾向和更好的稳定性,并显示了更强的毒性。
3 Aβ寡聚体a结构类型
与神经毒性有关的Aβ寡聚体存在不同的聚集状态,这些不同聚集状态的寡聚体在致病毒性方面可能存在显著的差别。寡聚体的二级结构主要是由平行与反平行的折叠混合构成的,而纤维状寡聚体的二级结构则是由平行的折叠堆垛状结构构成。在AD中发挥致病作用的淀粉样蛋白组装状态可能包括低分子量寡聚体(例如二聚体和三聚体)、较大球状寡聚体、淀粉样物质衍生的可扩散性配体(十二聚体,35~60 kDa)、淀粉样蛋白球体和球聚体(十二聚体,38~48 kDa)、环形原纤维(APFs,100~150 kDa)、原纤维和纤维体,还有包含密集Aβ纤维和大量其他分子和细胞成分的Aβ斑块。各种不同的因素都会影响Aβ寡聚体结构形成,这些因素包括不限于合成Aβ肽的种类及纯度、有无突变、溶液的肽浓度和离子浓度、环境的温度,以及酸碱度,同时体外条件下合成的寡聚体和体内提取的结构也存在很大的差异。对于小的Aβ寡聚体,细胞毒性随其尺寸增加升高,然而对于较大的Aβ寡聚体(12-mer或更高),细胞毒性随着尺寸增加而降低,表明对于中等大小的寡聚体细胞毒性可能是最大的。
Aβ二聚体水平在AD小鼠模型和AD患者中升高。Shankar等从AD脑中提取的可溶性Aβ寡聚体明显损害突触结构和功能,二聚体是其中最小的突触毒性寡聚体类型,二聚体可能是构成淀粉样斑块的基础结构或者可能由斑块内产生。AD脑皮质内的不溶性淀粉样斑块核心不损害LTP,除非它们被溶解释放出Aβ二聚体。这表明斑块核心大部分无活性,同时有效隔离了具有突触毒性的Aβ二聚体。Aβ二聚体可以组装形成更高分子量更稳定的结构,被认为是为毒性寡聚体提供结构单元的重要类型。三聚体是神经元胞外产生、分泌最多且最早的类型,Aβ三聚体很可能是组成Aβ 非纤维部分的基本结构,这可能是由于形成三聚体需要较低的能量或者由于缺乏分子伴侣不能转化为二聚体。当APP转基因大鼠3个月龄出现认知缺陷时,发现Aβ三聚体是神经元内唯一的Aβ寡聚体形式。在人体研究中发现三聚体早在1岁时就能被检测到,而二聚体直到50~60岁未在受试者体内检测到。
淀粉样物质衍生的可扩散性配体(ADDLs)被认为是一种可以在体外组装的 Aβ十二聚体,在Tg2576模型中,空间记忆未损害之前,ADDLs含量增加了5~100倍。与年龄匹配的对照组相比,ADDLs在AD脑组织和CSF中的含量增加70倍。这些表明ADDLs水平在AD中急剧上升。ADDLs诱导Fyn的激活,Fyn可以导致tau在18号酪氨酸位点过度磷酸化,三联体Aβ/ Fyn / tau在Aβ诱导与AD相关的认知损伤中发挥重要作用。ADDLs、球聚体可能代表不同的寡聚体组装形式,由ADDLs、球聚体抗体检测到的内源性Aβ种类的来源尚不清楚,需要进一步地研究来确定这些Aβ组装形式是如何产生的以及它们何时在AD脑内形成。
环形原纤维分子大小超过90 kDa时,它们类似于细菌成孔毒素,已经表明APFs通过渗透作用改变神经元脂质膜动态平衡来并诱导细胞死亡。
4 Aβ寡聚体损伤机制
Aβ寡聚体的损伤机制复杂,需要更多的研究来进一步阐明,可能涉及靶向突触抑制LTP、激活胶质细胞引发炎症反应、Ca2+稳态失衡、氧化应激、线粒体损伤、tau蛋白过度磷酸化、内质网应激、胰岛素抵抗和基因表达等多种机制。
Aβ寡聚体诱导的突触功能障碍依赖于N-甲基-D-天冬氨酸受体(NMDAR)的过度刺激,导致氧化应激的异常激活以及胞质Ca2+的升高,其反过来触发下游通路涉及磷酸化tau蛋白、胱氨酸天冬氨酸蛋白水解酶(caspases)、Cdk5/dynamin相关蛋白1(Drp1)、钙调磷酸酶/PP2B、PP2A、Gsk-3β、Fyn、cofilin和CaMKⅡ,并启动AMPA受体(AMPAR)以及NMDAR的内吞作用。多项研究表明Aβ寡聚体的毒性机制和酪氨酸蛋白激酶Fyn介导的信号中断有关。这些信号转导途径导致线粒体功能障碍,引发能量代谢紊乱和随后的突触功能障碍和突触丢失,以及LTP受损和认知能力下降。Aβ寡聚体作用于细胞膜受体NMDAR,通过细胞内信号转导致突触后膜致密体和突触囊泡转运中的PSD-95和MP降解(美金刚和MK-801可以阻断),神经细胞树突棘和突触减少,损害神经元突触可塑性,甚至诱导细胞凋亡。Aβ寡聚体可以激活特定的信号转导通路导致tau蛋白的异常磷酸化。Aβ寡聚体作用于原代皮质神经元中的NMDA受体(NMDAR),增加NMDAR依赖性的Ca2+内流,活化细胞内Ca2+浓度依赖性钙调蛋白激酶Ⅱα(CaMKⅡα),活化的CaMKⅡα诱导tau蛋白的异常磷酸化。
神经元内Aβ寡聚体可作用于3×Tg小鼠α7-神经型乙酰胆碱受体(α7nAChRs)以及LTP,导致认知缺陷,同时也有证据表明Aβ寡聚体可作为细胞外配体与受体结合作用于突触,影响神经细胞功能。Ferretti等研究神经炎症可能是Aβ寡聚体在细胞内积累引起的最早病理反应之一,神经元特异性环氧合酶2(COX-2)在AD疾病的早期被上调,此酶是由携带Aβ寡聚体的神经元细胞特异性表达。
Aβ寡聚体能与特异性受体结合使小胶质细胞激活、增殖,并分泌释放大量炎性细胞因子和神经毒性,从而引起中枢神经系统的损伤。White等在体外培养的星形胶质细胞中发现:Aβ寡聚体和纤维体均可引起IL-1β释放增加,但是寡聚体可明显增加NO和TNF-α的释放,而纤维体对两种炎性因子的释放无明显影响,提示寡聚体在引发星形胶质细胞介导的炎症反应中作用更强。
近来研究已经将Aβ寡聚体引发的神经病理性机制与糖尿病外周胰岛素抵抗相关的机制联系起来,由Aβ寡聚体诱导的胰岛素信号传导功能失调可能和AD发病有关。Aβ寡聚体的毒性作用可能会损害正常的大脑胰岛素信号传导以及导致葡萄糖和脂质的增加,并引发前向级联反应,通过增加细胞应激(例如胞浆钙超载、氧化应激、内质网应激)破坏神经元功能,反过来加剧了神经元胰岛素抵抗,导致Aβ的产生增多和Aβ寡聚体清除率的降低,这种恶性循环导致认知障碍和AD。
Laurén等通过克隆表达细胞朊蛋白(PrPc)确定其为Aβ寡聚体的受体,抗PrPc抗体可以阻断Aβ寡聚体与PrPc的结合,并逆转Aβ寡聚体对海马切片突触可塑性的损害。Aβ 寡聚体与小鼠PrPc结合持续损害海马LTP、记忆和学习功能。
最新提出的Aβ/Aβ寡聚体可能参与结合的受体已有16种之多,但还有单一的受体被证明在寡聚体的毒性作用中不可或缺。不同类型Aβ寡聚体可能通过不同途径发挥毒性作用,但是毒性作用具体是由哪一种或哪几种寡聚体引发尚不清楚。
5 靶向Aβ寡聚体的疾病修饰治疗
Aβ寡聚体可能是AD脑内发挥毒性作用的最重要的类型,其朊蛋白样播散作用可能是AD疾病进展的重要机制。Aβ寡聚体作为靶点的治疗方法越来越多地出现,其中靶向Aβ寡聚体的免疫治疗最有希望成为未来AD的疾病修饰疗法。目前有4种人源化单克隆抗体进入了3期临床试验,包括Solanezumab、Gantenerumab、Aducanumab、Lecanemab,其在减缓受试者认知损害和避免副作用方面均显示出一定的优势。总结这几个单克隆抗体结合类型,都是把Aβ寡聚体作为结合的主要方面。AβpE3是AD脑内重要的Aβ成分,由于毒性作用近来受到越来越多的重视。一种特异性结合AβpE3寡聚体的单克隆抗体9D5,免疫组化染色显示结合细胞内的Aβ同时较强的亲和血管壁上的Aβ成分,但是不结合老年斑,免疫治疗5×FAD转基因小鼠显示减少了脑内的Aβ40、Aβ42和AβpE3,老年斑显著减少,并改善了小鼠的认知功能。由于AD患者大多为老年人,可能未能通过主动免疫治疗产生足够的抗体来清除体内的Aβ,被动免疫治疗很好的弥补了这一缺陷。近来Aducanumab、Lecanemab先后获得了FDA批准用于轻度认知障碍或轻度痴呆期AD的治疗。Aducanumab结合抗原表位Aβ3-7,选择性结合Aβ寡聚体、纤维体和老年斑,在3期临床试验中显示出明显的清除Aβ效果,且高剂量组延缓了认知损害。Lecanemab结合抗原表位Aβ1-16,优先结合Aβ原纤维,同时结合并清除纤维体和老年斑,在3期临床试验中显示出清除Aβ负荷,减缓认知损害的作用。研究认为其关键特点就是具有结合神经毒性可溶Aβ寡聚体的能力,并且对Aβ单体具有较低的亲和力。由于靶向Aβ的被动免疫治疗会出现血管源性水肿和微出血的副作用,且清除Aβ类型的局限性,目前限制了其进一步的应用,亟需开发高度特异性靶向毒性Aβ并且不良反应轻微的药物,阻断AD疾病的进展。
6 待解决问题及展望
Aβ寡聚体和AD的临床相关性已经确立,需要进一步解决的是建立这些不同类型寡聚体在人和动物模型中的纵向分布,以及检测这些可溶性Aβ寡聚体含量与认知障碍之间的相对关系。多个体外和体内模型的实验阐明了Aβ寡聚体的广泛致病性,关于Aβ寡聚体如何靶向结合以及导致细胞损伤机制的研究有了显著的进展,与AD相关的Aβ寡聚体结构难题正在不断的通过新技术应用解决。由于致病性寡聚体在疾病早期出现并贯穿了AD的整个发病过程,这就为AD的诊断治疗提供了可行的途径,可以研发高特异性的中和抗体在疾病早期阻断并清除寡聚体,减小药物的副作用从而减少微出血和脑组织水肿,研发寡聚体探针核素标记物开启早期特异性的功能脑成像技术。
参考文献
[1]Alzheimer's A. 2016 Alzheimer's disease facts and figures[J]. Alzheimers Dement,2016,12(4):459-509.
[2]Hardy JA,Higgins GA. Alzheimer's disease:the amyloid cascade hypothesis[J]. Science,1992,256(5054):184-185.
[3]Herrup K. The case for rejecting the amyloid cascade hypothesis[J]. Nat Neurosci,2015,18(6):794-799.
[4]Holmes C,Boche D,Wilkinson D,et al. Long-term effects of abeta42 immunisation in Alzheimer's disease:follow-up of a randomised,placebo-controlled phase I trial[J]. Lancet(London,England),2008,372(9634):216-223.
[5]Haass C,Selkoe DJ. Soluble protein oligomers in neurodegeneration:lessons from the Alzheimer's amyloid beta-peptide[J]. Nat Rev Mol Cell Biol,2007,8(2):101-112.
[6]Gulisano W,Maugeri D,Baltrons MA,et al. Role of amyloid-beta and tau proteins in Alzheimer's disease:confuting the amyloid cascade[J]. Journal of Alzheimer's disease:JAD,2018,64(s1):S611-S631.
[7]Kim J,Onstead L,Randle S,et al. Abeta40 inhibits amyloid deposition in vivo[J]. J Neurosci,2007,27(3):627-633.
[8]Chia PZ,Gleeson PA. Intracellular trafficking of the beta-secretase and processing of amyloid precursor protein[J]. IUBMB Life,2011,63(9):721-729.
[9]Xia D,Watanabe H,Wu B,et al. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer's disease[J]. Neuron,2015,85(5):967-981.
[10]Shimada H,Ataka S,Tomiyama T,et al. Clinical course of patients with familial early-onset Alzheimer's disease potentially lacking senile plaques bearing the E693Delta mutation in amyloid precursor protein[J]. Dement Geriatr Cogn Disord,2011,32(1):45-54.
[11]Lesne SE,Sherman MA,Grant M,et al. Brain amyloid-beta oligomers in ageing and Alzheimer's disease[J]. Brain,2013,136(Pt 5):1383-1398.
[12]Koffie RM,Meyer-Luehmann M,Hashimoto T,et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques[J]. Proc Natl Acad Sci USA,2009,106(10):4012-4017.
[13]Wang ZX,Tan L,Liu J,et al. The essential role of soluble abeta oligomers in Alzheimer's disease[J]. Molecular Neurobiology,2016,53(3):1905-1924.
[14]Liao D,Miller EC,Teravskis PJ. Tau acts as a mediator for Alzheimer's disease-related synaptic deficits[J]. Eur J Neurosci,2014,39(7):1202-1213.
[15]Quintela-Lopez T,Ortiz-Sanz C,Serrano-Regal MP,et al. Abeta oligomers promote oligodendrocyte differentiation and maturation via integrin beta1 and Fyn kinase signaling[J]. Cell Death Dis,2019,10(6):445.
[16]Crestini A,Santilli F,Martellucci S,et al. Prions and neurodegenerative diseases:a focus on Alzheimer's disease[J]. Journal of Alzheimer's disease:JAD,2022,85(2):503-518.
[17]Lambert MP,Barlow AK,Chromy BA,et al. Diffusible,nonfibrillar ligands derived from abeta1-42 are potent central nervous system neurotoxins[J]. Proc Natl Acad Sci USA,1998,95(11):6448-6453.
[18]Walsh DM,Klyubin I,Fadeeva JV,et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo[J]. Nature,2002,416(6880):535-539.
[19]Klyubin I,Walsh DM,Lemere CA,et al. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo[J]. Nature Medicine,2005,11(5):556-561.
[20]Larson ME,Lesne SE. Soluble Abeta oligomer production and toxicity[J]. Journal of Neurochemistry,2012,120(Suppl1):125-139.
[21]Cheng IH,Scearce-Levie K,Legleiter J,et al. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models[J]. The Journal of Biological Chemistry,2007,282(33):23818-23828.
[22]Miravalle L,Calero M,Takao M,et al. Amino-terminally truncated abeta peptide species are the main component of cotton wool plaques[J]. Biochemistry,2005,44(32):10810-10821.
[23]Sofola-Adesakin O,Khericha M,Snoeren I,et al. pGluAbeta increases accumulation of abeta in vivo and exacerbates its toxicity[J]. Acta Neuropathologica Communications,2016,4(1):109.
[24]Jan A,Hartley DM,Lashuel HA. Preparation and characterization of toxic Abeta aggregates for structural and functional studies in Alzheimer's disease research[J]. Nat Protoc,2010,5(6):1186-1209.
[25]Stine WB,Dahlgren KN,Krafft GA,et al. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis[J]. The Journal of Biological Chemistry,2003,278(13):11612-11622.
[26]Yang T,Li S,Xu H,et al. Large soluble oligomers of amyloid beta-protein from alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate[J]. J Neurosci,2017,37(1):152-163.
[27]Mc Donald JM,Savva GM,Brayne C,et al. The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia[J]. Brain,2010,133(Pt5):1328-1341.
[28]Shankar GM,Li S,Mehta TH,et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory[J]. Nature Medicine,2008,14(8):837-842.
[29]Mc Donald JM,O'Malley TT,Liu W,et al. The aqueous phase of Alzheimer's disease brain contains assemblies built from approximately 4 and approximately 7 kDa abeta species[J]. Alzheimers Dement,2015,11(11):1286-1305.
[30]Leon WC,Canneva F,Partridge V,et al. A novel transgenic rat model with a full Alzheimer's-like amyloid pathology displays pre-plaque intracellular amyloid-beta-associated cognitive impairment[J]. J Alzheimers Dis,2010,20(1):113-126.
[31]Haass C,Mandelkow E. Fyn-tau-amyloid:a toxic triad[J]. Cell,2010,142(3):356-358.
[32]Lashuel HA,Lansbury PT. Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins[J]. Q Rev Biophys,2006,39(2):167-201.
[33]Cline EN,Bicca MA,Viola KL,et al. The Amyloid-beta oligomer hypothesis:beginning of the third decade[J]. J Alzheimers Dis,2018,64(s1):S567-S610.
[34]Jarosz-Griffiths HH,Noble E,Rushworth JV,et al. Amyloid-beta receptors:the bood,the bad,and the prion protein[J]. J Biol Chem,2016,291(7):3174-3183.
[35]Zhang C,Qiu HE,Krafft GA,et al. A beta peptide enhances focal adhesion kinase/Fyn association in a rat CNS nerve cell line[J]. Neurosci Lett,1996,211(3):187-190.
[36]Chin J,Palop JJ,Yu GQ,et al. Fyn kinase modulates synaptotoxicity,but not aberrant sprouting,in human amyloid precursor protein transgenic mice[J]. J Neurosci,2004,24(19):4692-4697.
[37]Tu S,Okamoto S,Lipton SA,et al. Oligomeric abeta-induced synaptic dysfunction in Alzheimer's disease[J]. Mol Neurodegener,2014,9:48.
[38]Decker H,Jurgensen S,Adrover MF,et al. N-methyl-D-aspartate receptors are required for synaptic targeting of Alzheimer's toxic amyloid-beta peptide oligomers [J]. Journal of Neurochemistry,2010,115(6):1520-1529.
[39]Amar F,Sherman MA,Rush T,et al. The amyloid-beta oligomer Abeta*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation[J]. Sci Signal,2017,10(478):Sci Signal,2017,10(478):2021.
[40]Wallace TL,Porter RH. Targeting the nicotinic alpha7 acetylcholine receptor to enhance cognition in disease [J]. Biochemical Pharmacology,2011,82(8):891-903.
[41]Ferretti MT,Bruno MA,Ducatenzeiler A,et al. Intracellular Abeta-oligomers and early inflammation in a model of Alzheimer's disease [J]. Neurobiology of Aging,2012,33(7):1329-1342.
[42]Sondag CM,Dhawan G,Combs CK. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia [J]. J Neuroinflammation,2009,6:1.
[43]De Felice FG,Lourenco MV,Ferreira ST. How does brain insulin resistance develop in Alzheimer's disease[J]. Alzheimers Dement,2014,10(1 Suppl):S26-S32.
[44]Viola KL,Klein WL. Amyloid beta oligomers in Alzheimer's disease pathogenesis,treatment,and diagnosis [J]. Acta Neuropathologica,2015,129(2):183-206.
[45]Laurén J,Gimbel DA,Nygaard HB,et al. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers[J]. Nature,2009,457(7233):1128-1132.
[46]Laurén J. Cellular prion protein as a therapeutic target in Alzheimer's disease[J]. Journal of Alzheimer's Disease:JAD,2014,38(2):227-244.
[47]Nussbaum JM,Schilling S,Cynis H,et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-beta[J]. Nature,2012,485(7400):651-655.
[48]Rasool S,Martinez-Coria H,Wu JW,et al. Systemic vaccination with anti-oligomeric monoclonal antibodies improves cognitive function by reducing abeta deposition and tau pathology in 3xTg-AD mice[J]. Journal of Neurochemistry,2013,126(4):473-482.
[49]van Dyck CH. Anti-Amyloid-beta monoclonal antibodies for Alzheimer's disease:pitfalls and promise[J]. Biological psychiatry,2018,83(4):311-319.
[50]Jawhar S,Wirths O,Bayer TA. Pyroglutamate amyloid-beta(Abeta):a hatchet man in Alzheimer disease[J]. The Journal of biological chemistry,2011,286(45):38825-38832.
[51]Piechotta A,Parthier C,Kleinschmidt M,et al. Structural and functional analyses of pyroglutamate-amyloid-beta-specific antibodies as a basis for Alzheimer immunotherapy[J]. J Biol Chem,2017,292(30):12713-12724.
[52]Wirths O,Erck C,Martens H,et al. Identification of low molecular weight pyroglutamate A{beta} oligomers in Alzheimer disease:a novel tool for therapy and diagnosis[J]. J Biol Chem,2010,285(53):41517-41524.
[53]Arndt JW,Qian F,Smith BA,et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-beta[J]. Sci Rep,2018,8(1):6412.
[54]Budd Haeberlein S,O'Gorman J,Chiao P,et al. Clinical development of aducanumab,an anti-abeta human monoclonal antibody being investigated for the treatment of early Alzheimer's disease[J]. J Prev Alzheimers Dis,2017,4(4):255-263.
[55]van Dyck CH,Swanson CJ,Aisen P,et al. Lecanemab in early Alzheimer's disease[J]. N Engl J Med,2023,388(1):9-21.
[56]Linse S,Scheidt T,Bernfur K,et al. Kinetic fingerprints differentiate the mechanisms of action of anti-abeta antibodies[J]. Nat Struct Mol Biol,2020,27(12):1125-1133.
[57]Sevigny J,Chiao P,Bussiere T,et al. The antibody aducanumab reduces abeta plaques in Alzheimer's disease[J]. Nature,2016,537(7618):50-56.
[58]Cehlar O,Skrabana R,Revajova V,et al. Structural aspects of Alzheimer's disease immunotherapy targeted against amyloid-beta peptide[J]. Bratisl Lek Listy,2018,119(4):201-204.

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言