黏多糖贮积症Ⅳ型:症状与体征、病因、流行病学、诊断与治疗
2022-08-24 MedSci原创 MedSci原创

黏多糖贮积症Ⅳ型,又称莫尔奎(Morquio)综合征、骨软骨营养不良、畸形性软骨营养不良、非典型性佝偻病。其特征性表现为躯干明显变短,矮小畸形,股骨头和髋臼呈进行性改变,关节肿大,肌肉韧带松弛。本病为
黏多糖贮积症Ⅳ型,又称莫尔奎(Morquio)综合征、骨软骨营养不良、畸形性软骨营养不良、非典型性佝偻病。其特征性表现为躯干明显变短,矮小畸形,股骨头和髋臼呈进行性改变,关节肿大,肌肉韧带松弛。本病为常染色体隐性遗传,男稍多于女。
一、一般概述
粘多糖贮积症 IV (MPS IV) 是一种粘多糖贮积病,以两种形式存在(MPS IVA 和 MPS IVB)。这些是常染色体隐性遗传病,包括一个连续体,由一种快速发展的严重形式和另一种缓慢发展的形式组成。严重的形式在 1 到 3 岁之间变得明显,通常表现为膝关节和胸骨突出。缓慢进展的形式可能直到青春期才变得明显,表现为髋部疼痛和僵硬。
尽管它们是同一疾病的亚型,但为了有效治疗和疾病管理,区分 MPS IVA 和 MPS IVB 很重要。 MPS IVA 与 MPS IVB 只能通过分子遗传学或生化测试来区分,因为它们呈现的症状相似。 MPS IV 的发生是由于 N-乙酰基-半乳糖胺-6-硫酸酯酶 (GALNS) 酶的缺乏而发生的,而 MPS IVB 是由于 β-半乳糖苷酶的缺乏而发生的。
任何一种酶的缺乏都会导致体内粘多糖的积累、骨骼发育异常和其他症状。在大多数情况下,患有 MPS IV 的人智力正常。 MPS IVB 的临床特征通常比 MPS IVA 相关的临床特征更少且更温和。酶替代疗法可用于治疗 MPS IVA。
粘多糖贮积症(MPS)是一组遗传性溶酶体贮积症。溶酶体作为细胞内的主要消化单位发挥作用。溶酶体中的酶分解或消化特定的营养物质,例如某些碳水化合物和脂肪。在患有 MPS 疾病的个体中,特定溶酶体酶的缺乏或功能障碍会导致某些复杂碳水化合物(粘多糖或糖胺聚糖)在动脉、骨骼、眼睛、关节、耳朵、皮肤和/或牙齿中异常积累。这些积累也可能存在于呼吸系统、肝脏、脾脏、中枢神经系统、血液和骨髓中。这种积累最终会对身体的细胞、组织和各种器官系统造成渐进性损伤。粘多糖贮积症有几种不同的类型和亚型。
二、症状与体征
MPS IV 的症状可能包括生长迟缓;下脸突出;脊柱的侧向和前后或左右弯曲异常(脊柱侧凸)或担心脊柱异常;脖子异常短;膝盖异常靠近(敲膝或膝外翻);扁平足;长骨(骨骺)生长端发育异常;髋关节脱位和关节炎和/或突出的胸骨(鸡胸)。也可能发生听力丧失、腿部无力和/或其他异常。
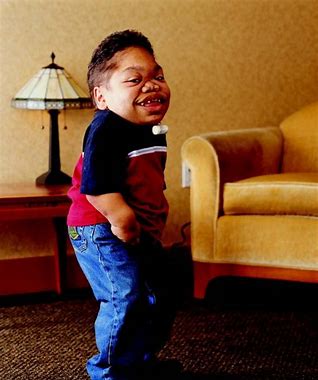
受影响的儿童具有特征性的面部外观,可能包括扩大的头部、宽阔的嘴巴、突出的颧骨、异常小的鼻子、间距宽且釉质薄的牙齿,以及眼睛大而分散并伴有轻微的角膜混浊。肝脏和脾脏可能会轻度肿大。患有 MPS IV 的儿童在生命早期就表现出明显的生长异常,躯干短而四肢正常。肘、腕、髋、膝等大关节异常灵活,造成整体不稳定。受影响的个体表现出经常跌倒的蹒跚步态。与其他 MPS 贮积症不同,早期发育和智力通常是正常的。高频听力障碍很常见。

骨骼 X 射线通常显示椎骨明显变平。胳膊和腿的长骨比正常人更短更厚。对于身体的其他部分来说,头骨很大。颈部第一和第二椎骨之间的连接发育不良,这种异常可能危及生命。轻微的损伤可能会导致两个椎骨相互滑动并压迫脊髓。通常通过脊柱融合术来稳定上颈椎的手术可以挽救生命,但尽管进行了手术,但预期寿命会有所降低。胸部的畸形会对心脏和肺部造成压力,最终可能导致呼吸衰竭。
三、病因
MPS IVA 是一种常染色体隐性遗传病,是由于 GALNS 基因突变导致 GALNS 酶缺乏引起的。
MPS IVB 是一种常染色体隐性遗传疾病,由 GLB1 基因突变导致的 β-半乳糖苷酶缺乏引起。
两者都会导致硫酸角质素 (KS) 在身体的细胞和组织中积累。 KS 在角膜和骨骼中的积累分别导致视力下降和骨骼畸形。
遗传疾病是由来自父亲和母亲的染色体上特定性状的基因组合决定的。隐性遗传疾病发生在个体遗传了同一性状的异常基因的两个拷贝时,每个拷贝来自父母一方。如果一个人接受了一种正常基因和一种疾病基因,则该人将成为该疾病的携带者,但通常不会出现症状。每次怀孕,两个携带者父母都通过缺陷基因并有受影响的孩子的风险是 25%。每次怀孕生下一个像父母一样是携带者的孩子的风险是 50%。一个孩子从父母双方那里获得正常基因并在该特定特征上遗传正常的机会是 25%。男性和女性的风险相同。
所有个体都携带 4-5 个异常基因。近亲(近亲)的父母比无关父母携带相同异常基因的可能性更高,这增加了患隐性遗传疾病的孩子的风险。
四、流行病学
MPS IV 对男性和女性的影响相同。 流行率的估计范围从 1/40,000 到 1/200,000 出生。 MPS IVA(95% 的受 MPS IV 影响的个体)比 MPS IVB(5% 的受影响个体)更常见。
作为一个群体,溶酶体贮积病(MPS 是其中的一个亚组)据信估计频率约为每 5,000 名活产婴儿中的一名。 尽管个别疾病很少见,但该群体共同影响着世界各地的许多人。
五、鉴别诊断
GLB1 基因的突变也可能与另一种称为 GM1 神经节苷脂沉积症的溶酶体贮积病有关,这是一种常染色体隐性遗传疾病,会逐渐破坏大脑和脊髓中的神经细胞。
MPS IVA 和 MPS IVB 的体征和症状与其他类型的 MPS 贮积病和粘脂症重叠,这是一个类似疾病家族,其症状与 MPS 贮积病非常相似。有关这些情况的信息可以在罕见病数据库中找到。
脊柱骨骺发育不良 (SED) 是一种罕见的遗传性骨骼疾病,仅影响男性,X 线检查结果与 MPS IVA 患者相似,但也存在其他特征。身体特征包括中度身材矮小(侏儒症)、中度至重度脊柱畸形、桶形胸部、不成比例的短躯干和过早的骨关节炎。
如果该病最初仅表现为髋部疼痛,则可能会误诊 MPS IVA 患者的 Legg-Calve-Perthes 病。
六、诊断
病史、体格检查、骨骼 X 射线和尿糖胺聚糖 (GAG) 分析的结果提示 MPS IV 诊断。 尿液中通常会出现过量的硫酸角质素。
MPS IVA 的诊断通过培养的血液或皮肤细胞中的低 GALNS 酶活性和/或用于识别 GALNS 基因突变的分子遗传学检测来证实。
MPS IVB 诊断通过在血液或皮肤细胞中发现 β-半乳糖苷酶缺乏和/或通过分子遗传学检测来识别 GLB1 基因突变来确认。
七、治疗
2014 年,FDA 批准了一种重组人 GALNS 酶替代疗法(elosulfase alfa,或 Vimizim)用于治疗 MPS IVA。 Vimizim 由 BioMarin Pharmaceutical Inc. 制造。详细见:NICE推荐Vimizim用于罕见病-4A型粘多糖贮积症
MPS IV 的其他治疗是对症治疗和支持治疗。将上颈部的骨头减压并融合到颅底的手术可以防止颈椎的不稳定和对脊髓的潜在损伤。
MPS IV 受影响个体的管理最好由多名专家进行,包括:物理康复治疗师、心理支持精神科医生、学习优化教育专业人员以及医疗设备依赖受影响个体的家庭护理专业人员。
外科医生也可能在治疗受影响的个体方面发挥关键作用。对于患有心室肥大(过度生长)的受影响的 induvial,可能需要放置生物瓣膜或人工瓣膜。可能需要切除增大的扁桃体和腺样体,以缓解上气道阻塞和睡眠呼吸暂停。此外,有听力损失的人可能需要通气管和助听器。可能需要穿透性角膜移植术(角膜置换术)来治疗导致视力受损的角膜混浊(角膜瘢痕或混浊)。
由于患有 MPS IVA 的儿童智力正常,他们通常参加常规课程,但如果他们有听力或视力障碍,他们需要坐在靠近教室前面的位置。他们可能还需要在校园周围使用轮椅。
建议对受影响的个人及其家人进行遗传咨询。
详细见:黏多糖贮积症ⅣA型诊治共识
八、预后
可活到成人。多死于心肺功能衰竭。多数病人死于20岁之前。
九、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
参考资料:
Hendriksz C, Burton BK, Fleming T, et al.. A multi-national, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of BMN 110 treatment for mucopolysaccharidosis IVA (Morquio syndrome type A). Mol Genet Metab. 2013a;108:S48.
Tomatsu S, et al. Mucopolysaccharidoses IVA (Morquio A): identification of novel common mutations in the N-acetylgalactosamine-6-sulfate sulfatase (GALNS) gene in Italians patients. Hum Mutat. 2004;24:187-8.
Walker PP, Rose E, Williams JG. Upper airways abnormalities and tracheal problems in Morquio’s disease. Thorax. 2003;58:458-9.
Kjellen L, Lindahl V. The proteoglycans structures and functions. Ann Rev Biochem 1991;60:443-475.
Regier DS, Oetgen M, Tanpaiboon P. Mucopolysaccharidosis Type IVA. 2013 Jul 11 [Updated 2016 Mar 24]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK148668/ Accessed August 21, 2019.
McKusick VA, ed. Online Mendelian Inheritance in Man (OMIM). Baltimore, MD: The Johns Hopkins University; Entry No. 253000; Available at http://omim.org/entry/253000 Last Update: 05/31/2018. Accessed August 21, 2019.
McKusick VA, ed. Online Mendelian Inheritance in Man (OMIM). Baltimore, MD: The Johns Hopkins University; Entry No. 25301; Available at http://omim.org/entry/253010 Last Update: 07/09/2016. Accessed August 21, 2019.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言









#诊断与治疗#
162
#流行病#
60