JMC:新型选择性强效靶向ERα的PROTAC降解剂,体内外有效克服乳腺癌激素治疗耐药
2023-05-18 精准药物 网络 发表于上海

新型选择性强效靶向ERα的PROTAC降解剂,体内外有效克服乳腺癌激素治疗耐药。
乳腺癌是严重威胁人类身体健康的一类疾病,其中大约80%的乳腺癌患者表现为雌激素受体阳性(ER+)。目前,在临床上,针对ER+乳腺癌患者的治疗方式主要是内分泌疗法,包括选择性ER调控剂(SERMs)以及选择性ER降解剂(SERDs)(如下图)。
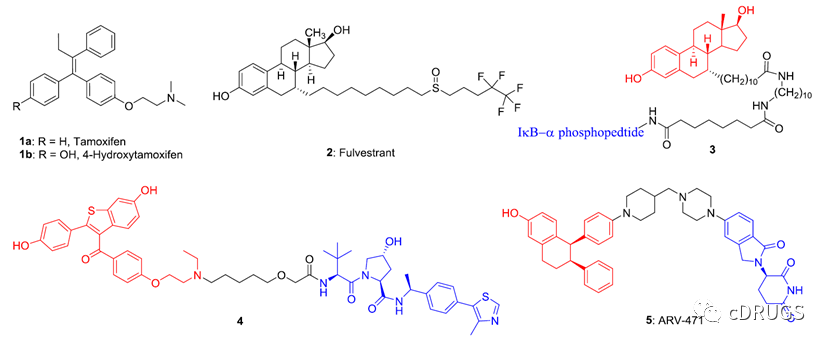
然而,大约40%的乳腺癌患者在接受长期的Tamoxifen治疗后,将面临着获得性抗性,这将限制SERM药物Tamoxifen的治疗范围。对于SERD类药物,其能够降解ERα,导致ERα的减少,抑制ER信号通路进而抑制乳腺癌细胞的生长,这将在一定程度上减轻长期内分泌治疗带来的药物抗性。Fulvestrant是首个被FDA批准的用于治疗转移性ER+乳腺癌的SERD类药物,只是该药物的成药性差,不方便给药。2023年,Elacestrant是首个FDA批准的口服SERD类药物,用于治疗晚期或转移性乳腺癌患者。尽管这些SERD已经显示出相当大的临床益处,但对于ER+乳腺癌来说,内在和获得性耐药风险的增加仍然是一个重大挑战。最近,PROTAC技术发展迅速,多种靶向ER的PROTAC降解剂被开发出来,尽管ARV-471是临床进展最快的靶向降解ER的PROTAC分子,仍然只有少量用于治疗内分泌抗性乳腺癌细胞的PROTAC分子的体内研究被报道。此外,在人体中,ER包含两种亚型,ERα和ERβ,ERβ主要在良性组织中表达,抑制ER+乳腺癌细胞的增殖,而ERα对ER+乳腺癌细胞的增殖,分化以及侵袭起着重要的作用。因此,如何开发更精准的靶向ERα治疗ER+乳腺癌的药物仍然是很大的挑战。
近期,发表在Journal of Medicinal Chemistry 杂志上的“ Discovery of a Novel Class of PROTACs as Potent and Selective Estrogen Receptor α Degraders to Overcome Endocrine-Resistant Breast Cancer In Vitro and In Vivo ”报道了一种能够通过选择性降解ERα克服激素治疗抗性乳腺癌的PROTAC分子。
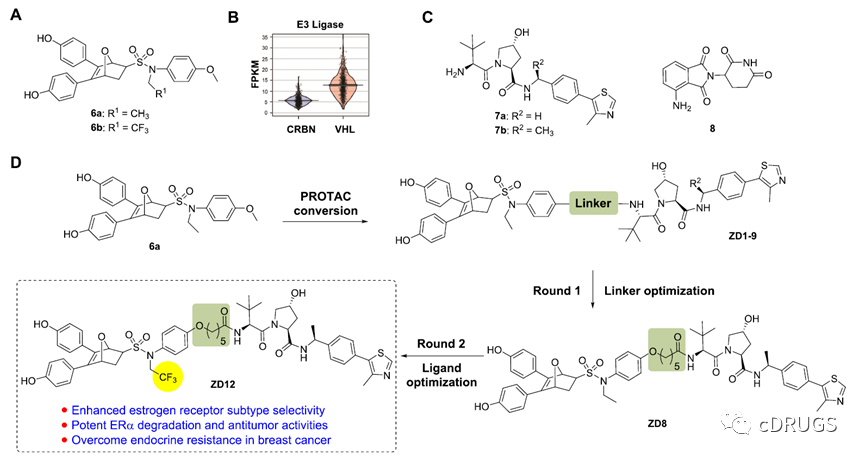
作者首先确定了一种新型ER药效团骨架—7-氧沙比环[2.2.1]庚烷磺酰胺(OBHSA)(如图,6a)作为ER蛋白招募配体。另外,在乳腺癌患者体内,VHL的表达显著高于CRBN,因此,选用VHL作为E3连接酶设计PROTAC可能更有利于获得具有高活性的PROTAC分子。同时,作者还设计并合成了一类基于CRBN的PROTAC分子作为比较。(所得化合物结构如下图所示)

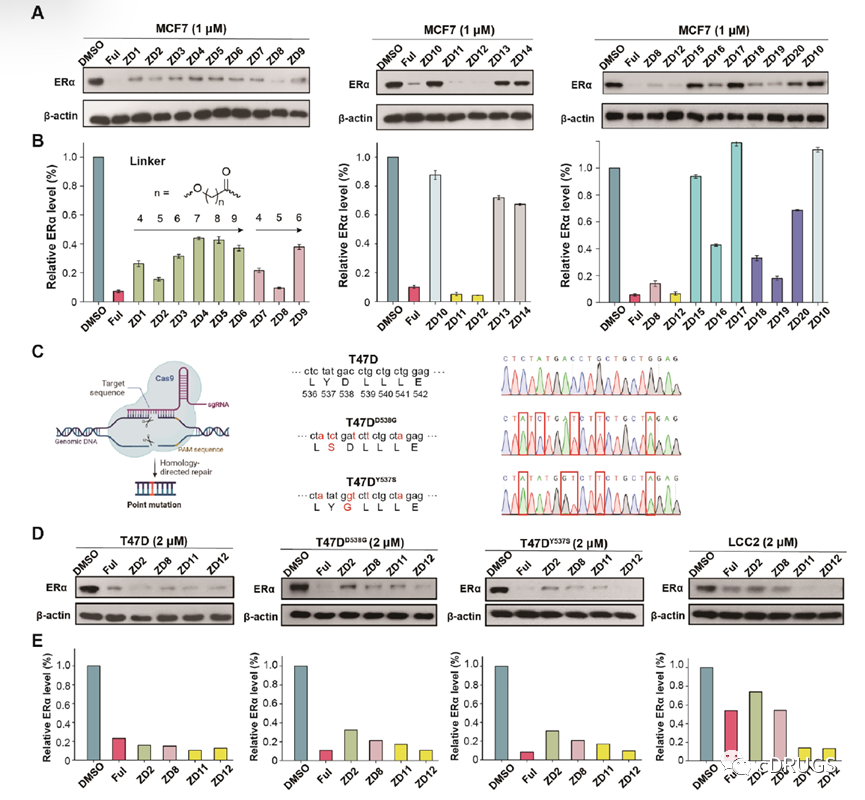
接下来,作者测试了ZD1-20的ERα降解活性(如图),结果表明,在MCF-7细胞中,多数化合物在1 μM浓度下对ERα表现出较强的降解活性,其中VHL招募的ZD2,ZD8,ZD11,ZD12在1 μM浓度下表现出和Fulvestrant相当甚至更佳的ERα选择性降解能力。此外,考虑到ERα突变(Y537S和D538G)是临床上报道的内分泌耐药的主要原因之一,作者构建了ERα突变的乳腺癌细胞系T47D D538G和T47D Y537S并发现优选化合物在2 μM浓度下不仅能够降解野生型ERα还能有效降解突变型ERα,并且在对Tamoxifen耐药的MCF-7细胞系LCC2中也能表现出显著的降解效果,这些结果表明优选化合物具有克服内分泌耐药的潜力。
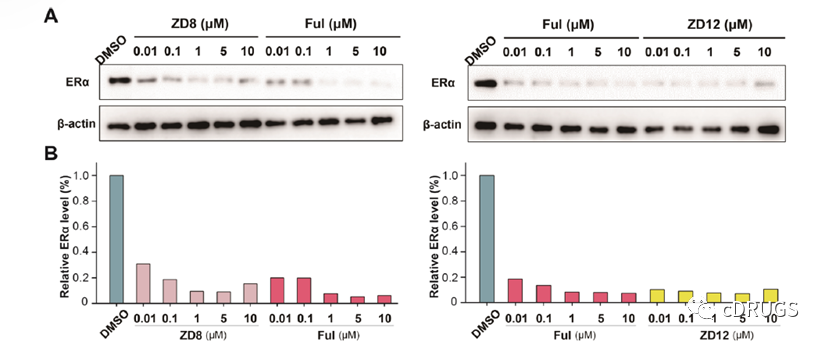
之后,作者选择了化合物ZD8和ZD12进行后续研究,并且发现这两种化合物在0.01到10 μM浓度下均能够实现ERα的有效降解,然而在浓度达到10 μM时,ERα蛋白质水平有一定程度上的恢复,这与Hook效应有关。并且,在相同浓度下ZD12的降解活性优于化合物Fulvestrant和ZD8。
作者进一步评估了在ERβ过表达细胞系NCI-H1975中ZD8和ZD12对ERβ的降解能力,发现这两种化合物在高浓度下不会实现ERβ的降解。鉴于PROTACs的降解机制,作者认为这种降解现象是由于ERα和ERβ蛋白在三元配合物形成过程中的结构差异,导致不同的ER亚型选择性降解活性。此外,作者还应用分子模型模拟了三元配合物的形成和选定的化合物ZD8与ERα-VHL蛋白复合物对接,结果表明,化合物ZD8能与ERα和VHL蛋白形成稳定的三元复合物。

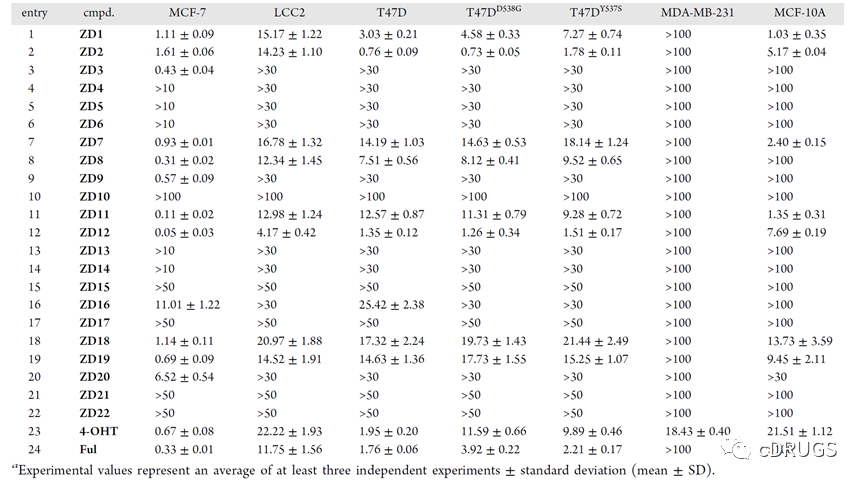
然后,作者以4-OHT和Ful作为阳性对照,测试了这些PROTACs对一系列乳腺癌细胞和正常乳腺细胞系的抑制活性(如表)。值得注意的是,这类化合物对三阴性乳腺癌细胞MDA-MB-231没有表现出抗增殖活性,说明该类化合物是通过降解ERα起到抗增殖活性的。另外,在对Tamoxifen敏感的MCF-7细胞中,ZD12的抑制活性优于4-OHT和Fulvestrant。重要的是,以ZD2和ZD12为代表的PROTACs对野生型和突变型ERα T47D细胞都表现出明显的抑制作用。
作者还探究了化合物ZD8和ZD12对ERα靶基因的影响,发现其能够抑制ERα靶基因新生RNA的合成并且ZD8和ZD12可以在全基因组范围内显著降低与ERα的结合,降低ERα靶基因表达。
最后,作者对优选化合物的体内抗肿瘤活性进行了探究。优选化合物ZD8,ZD12和ZD19均表现出适度的口服生物利用度,其中,ZD8以单次腹腔注射10 mg/kg的剂量表现出良好的药代动力学特性。在MCF-7异种移植小鼠体内,化合物ZD8表现出和Tamoxifen与Fulvestrant相当的肿瘤生长抑制作用,并且随着时间的延长,在相同剂量下,ZD8与Fulvestrant表现出优于Tamoxifen的抗肿瘤活性,在此期间没有观察到死亡和明显的体重下降。此外,在Tamoxifen耐药的ER+乳腺癌LCC2小鼠模型体内,ZD8和ZD12也表现出较强的抗肿瘤活性,并且在相同剂量下,ZD12在肿瘤组织中表现出更好的肿瘤抑制作用和ERα降解作用。

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言