NEJM:基因治疗将脊髓性肌萎缩症获重大突破
2017-11-03 MedSci MedSci原创

脊髓性肌萎缩症是致命的遗传性疾病,即使进行治疗,绝大多数患儿也活不过20个月。SMA此前被视为无药可医的遗传病,虽然发病率约为万分之一,但以中国每年出生1500万新生儿计算,国内目前的SMA患儿预计有3-5万名。 SMA患者体内,一种名为SMN的蛋白质因编码基因SMN-1的功能缺失而异常,而SMN蛋白对运动神经元的存活至关重要。没了这种蛋白,神经元无法正常工作,就导致了肌无力,肌肉迟缓、
脊髓性肌萎缩症是致命的遗传性疾病,即使进行治疗,绝大多数患儿也活不过20个月。SMA此前被视为无药可医的遗传病,虽然发病率约为万分之一,但以中国每年出生1500万新生儿计算,国内目前的SMA患儿预计有3-5万名。
SMA患者体内,一种名为SMN的蛋白质因编码基因SMN-1的功能缺失而异常,而SMN蛋白对运动神经元的存活至关重要。没了这种蛋白,神经元无法正常工作,就导致了肌无力,肌肉迟缓、萎缩,呼吸肌麻痹等多种症状的出现。针对这种机制,百健和Ionis制药联合开发的新药Spinraza于2016年12月获得FDA正式批准上市,SMA治疗专用药的空白才终于被填补。临床试验结果显示,该药通过对SMN-2基因的功能进行强化,增加SMN蛋白的数量,从而缓解疾病症状,使SMA患儿的死亡风险显著下降。
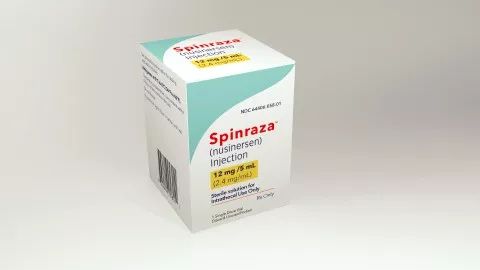
Spinraza目前仍是SMA患者治疗的唯一专用药物
这个药物的载体采用腺相关病毒载体(AAV)9型病毒,因为AAV9型病毒能穿透血脑屏障.

最新的临床3期结果显示,73名婴儿中,31名(51%)取得非常明显的疗效,而对照组为0%, P<0.001。同时可以降低无事件生存的风险为0.53(P=0.005),降低总死亡风险67%,同时显示出良好的安全性。
接受治疗的所有15名患儿生存期都跨过了20个月大关,而此前的研究显示能活到20个月的SMA患儿只有8%;15名患儿中只有8名需要面罩辅助呼吸;接受高剂量(低剂量3倍)基因治疗的12名患儿中,11名都可独立坐起至少5秒钟,正常吞咽进食,而除了Evelyn之外,还有一名小男孩Matteo也实现了独立走路,Matteo甚至能短暂跑动!这对于SMA患儿来说是难以想象的,此前由于缺乏特效疗法,他们大多只能终日躺下,靠呼吸机等手段维持生命。Mendell博士表示:“我从来没见过基因治疗能在一种致命疾病上取得如此好的效果。”

15名患儿生存期与SMA患者整体生存率的对照,基因治疗效果明显更好
当然,也有科学家对基因治疗的长期效果表示了怀疑,毕竟本次试验的随访时间到目前为止也只有两年多,还无法对基因治疗的长期效果提前下断言。
如果进一步的研究和试验证明了该疗法的长期可行性和普适性,基因治疗将开启一个全新的时代,因为已有众多的遗传病被明确了大脑内的相关基因,而AAV9载体则是首个在临床试验中对中枢神经系统投递基因效果如此之好的载体,使用极大的病毒量取得治疗成功也是史无前例的。而且,由于AVXS-101基因疗法的靶点是神经元,因此补充的基因不会因为细胞分裂而作用逐渐减弱,应当可以维持多年的效力。
原始出处:
Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, Chiriboga CA, Saito K, Servais L, Tizzano E, Topaloglu H, Tulinius M, Montes J, Glanzman AM, Bishop K, Zhong ZJ, Gheuens S, Bennett CF, Schneider E, Farwell W, De Vivo DC; ENDEAR Study Group. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med. 2017 Nov 2;377(18):1723-1732.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







#萎缩#
82
#脊髓性肌萎缩#
58
#肌萎缩#
65
#髓性#
70
厉害啊
112
很好的文章.谢谢分享
141