
拼命吃还是很瘦,日渐乏力,以为是营养不良,没想到竟是罕见病作祟!
11小时前 梅斯罕见新前沿 MedSci原创 发表于陕西省

26 岁李先生十年来四肢肌肉逐渐 “消失”,曾被诊为 “肌营养不良”,近期病情加重后确诊为晚发型庞贝病。文章介绍庞贝病,包括分型、表型、诊断要点和治疗方法。
26岁的李先生,十年来一直被一种奇怪的症状折磨:无论怎么吃,他的四肢肌肉却在逐渐“消失”。最初,他只是觉得手臂和大腿有些无力,但随着时间推移,这种乏力感越来越明显,甚至连提重物、爬楼梯都变得困难。他以为是营养不良或锻炼不足,试图通过补充营养和加强运动来改善,但却毫无起色。
8年前,他在外院检查时被诊断为“肌营养不良”,但因病情进展缓慢,他没有进一步治疗。直到近半年,肌无力明显加重,甚至运动时出现气促,他才意识到问题的严重性,再次求医。
真相大白
在全面检查中,李先生的身体特征和实验室数据逐渐勾勒出一个明确的诊断方向。虽然一般生命体征正常,但他体型消瘦,呈慢性病容,四肢肌肉明显萎缩,尤其是近端肌肉力量减弱。神经系统检查显示腱反射消失,而感觉系统正常,这与单纯的营养不良或神经性疾病并不一致。
进一步的实验室检查发现,李先生的肌酸激酶、谷草转氨酶和乳酸脱氢酶显著升高,提示肌肉损害;血乳酸水平运动后显著升高,但恢复较慢,表明代谢异常的可能性。心电图显示不完全性右束支传导阻滞和左心室高电压,心脏彩超提示部分心腔缩小,这些心脏表现引起了医生的警觉。
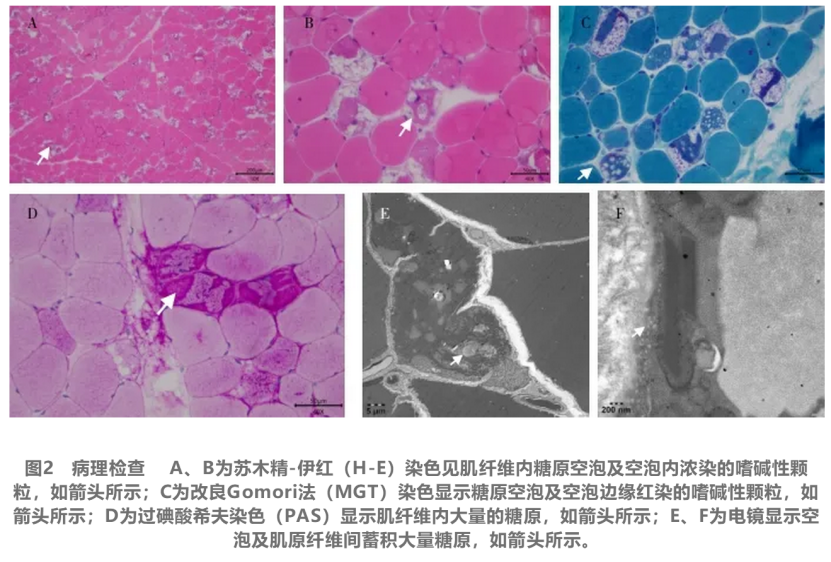
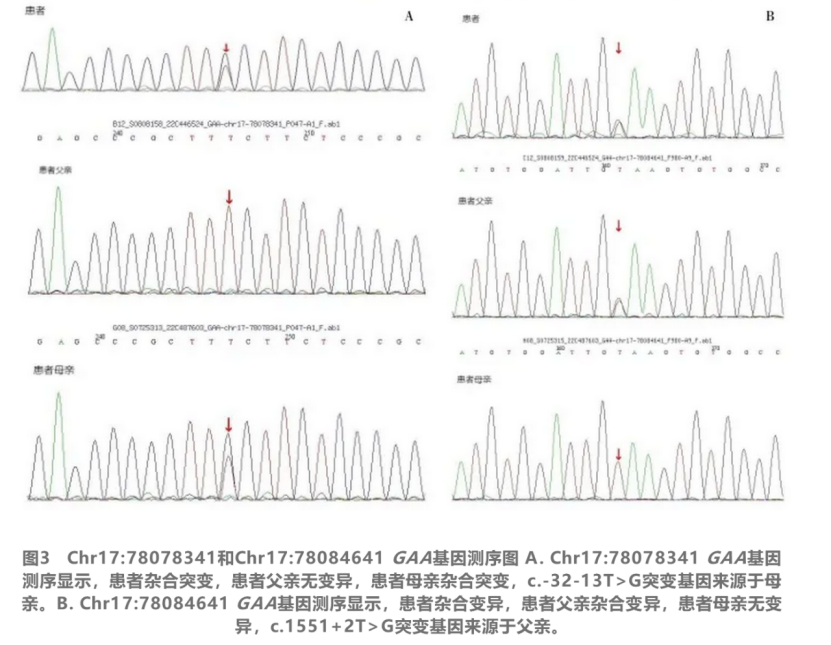
决定性的诊断来自肌肉活检和基因检测。肌肉病理检查发现肌纤维内存在大量糖原空泡,并通过多种染色技术和电镜进一步证实为糖原沉积。外周血酶学检测显示酸性α-葡萄糖苷酶活性严重不足,而基因测序揭示了GAA基因上的致病性突变。结合患者的临床表现、病理改变、酶学检测和遗传学证据,最终确诊为晚发型庞贝病。
什么是庞贝病?
庞贝病(PD)是一种罕见的常染色体隐性遗传病,其特征是酸性α-葡萄糖苷酶水平降低或缺失造成溶酶体降解糖原受损。庞贝病有两种主要分型:早发型庞贝病(IOPD)没有残留酶活性,常于婴儿期发病;晚发型庞贝病(LOPD)存留一定的酶活性,一般发病在儿童期、少年期或青年期。
多样表型与诊断要点
晚发型庞贝病(LOPD)具有多样的临床表型,即使在相似的遗传背景下,患者的表现也可能因营养、环境等非遗传因素而有所不同。典型的临床特征包括:
-
肌肉受累顺序
最常见的是下肢近端肌肉和轴肌受累,随后可能累及上肢和呼吸肌,表现为渐进性近端肌无力和运动耐力下降。
-
心脏和呼吸表现
少数患者可能出现心脏扩大或呼吸功能不全,但这并非所有患者的必备特征。
-
实验室与电生理检查
肌酸激酶(CK)升高:约95%的患者存在血清CK升高,通常为正常值上限的5倍以内。
电生理检查:常规神经传导多为正常,但针极肌电图约70%的患者显示肌病模式。
-
影像与病理诊断
核磁共振(MRI):定量肌肉MRI对评估肌肉受累程度具有重要价值。
肌肉病理:经典改变为空泡性肌病,但肌肉活检结果正常也不能完全排除庞贝病。
-
确诊方法
酸性α-葡萄糖苷酶活性检测是关键:酶活性低于参考范围提示诊断可能。
DNA分析进一步确诊:GAA基因的两种致病性突变可明确诊断。
治疗
重组人类GAA替代治疗是GSDⅡ的主要治疗方法。国外使用重组人类GAA替代治疗研究颇多,因GSDⅡ的酶替代制剂重组人类GAA国内上市较晚,国内相关研究较少见。婴儿型GSDⅡ使用重组人类GAA替代治疗治疗效果显著,可显著延长患儿生存期、改善心脏功能、运动功能及呼吸肌功能。
参考资料
1. 黄丽,张娇贵,陈定邦,等.糖原贮积病Ⅱ型1例[J].中国神经精神疾病杂志,2024,50(10):602-604.
2. 徐玲玲,巴宏军,李素萍,等.婴儿型糖原贮积症Ⅱ型酶替代治疗三例并文献复习[J].中国全科医学,2019,22(27):3377-3382.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言










#肌肉萎缩# #庞贝病#
6