综述:靶向EGFR的PROTAC类药物研发进展
2022-10-12 精准药物 精准药物

众所周知,PROTAC技术在克服耐药性及针对不可成药靶点具有极大优势,目前已经报道了多种针对EGFR的PROTAC,并在临床前研究中都表现较好的效果。
肺癌是世界上发病率和死亡率最高的恶性肿瘤,每年死亡人数可以达到140万至160万,占所有恶性肿瘤死亡人数的18%,具有高病发率、高死亡率的特点。其中,非小细胞肺癌 (NSCLC) 是最常见的肺癌类型(约75%的病例)。研究发现EGFR过表达与包括NSCLC、乳腺癌、胃癌等人类癌症疾病相关。目前已有三代EGFR抑制剂获批上市,极大地缓解了肺癌患者的痛苦,改善了患者的生活质量,但EGFR突变等引起的耐药性问题接踵而至。众所周知,PROTAC技术在克服耐药性及针对不可成药靶点具有极大优势,目前已经报道了多种针对EGFR的PROTAC,并在临床前研究中都表现较好的效果。
肺癌仍然是全球癌症死亡的主要原因,全球每年约有160万人死于肺癌。根据疾病类型,肺癌可分为小细胞肺癌(SCLC)或非小细胞肺癌(NSCLC)。其中,NSCLC占所有肺癌病例的85%,包括腺癌、鳞状细胞癌和大细胞癌。据报道,超过50% 的肺癌患者被诊断为晚期,而晚期非小细胞肺癌患者尚无治愈方法,使肺癌成为全球最具危害性的癌症类型。在过去的几十年里,化疗一直是晚期非小细胞肺癌患者的主要治疗选择,但治疗效果有限,且具有较大的副作用。
1、EGFR信号通路及其致病机制
表皮生长因子受体EGFR是一种跨膜蛋白,是ErbB家族的受体酪氨酸激酶 (RTK) 成员。在上皮细胞中,EGFR负责执行基本生命活动。研究表明,EGFR在许多人类恶性肿瘤中常见并过度表达。EGFR在人上皮细胞的信号转导起关键作用,参与细胞增殖、侵袭、凋亡和血管生成。EGFR由28个外显子和27个内含子组成,编码1186个氨基酸,其糖基化单体分子量约为170 kDa。EGFR的结构包括三个组成部分:细胞外结构域(两个配体结合结构域和两个富含半胱氨酸结构域),跨膜结构域和细胞内结构域(酪氨酸激酶结构域和具有多个自磷酸化位点的羧基末端尾部)。当外源性特异性配体与细胞外区域结合,发生EGFR活化和构象变化,进一步二聚化影响酪氨酸激酶活性并触发特定酪氨酸残基的自磷酸化,进而刺激信号级联和消耗ATP,引发疾病产生。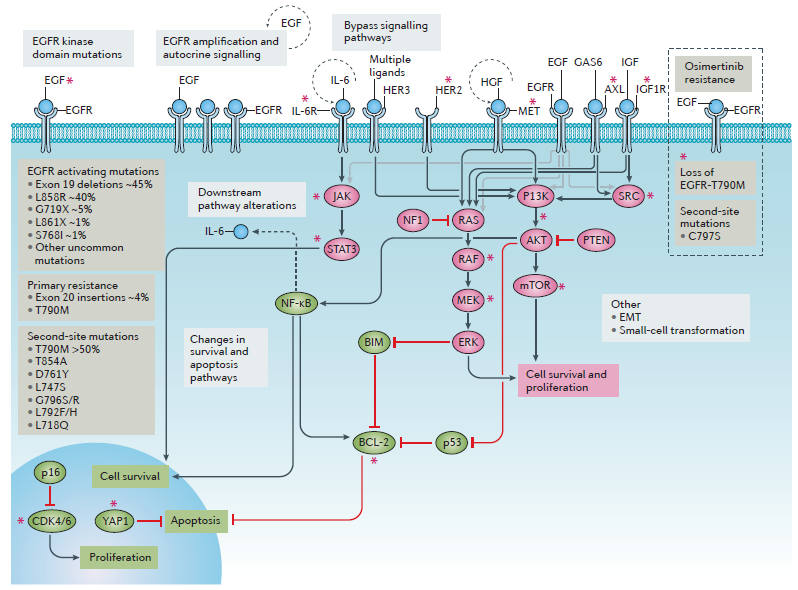
图1. EGFR信号通路及致病机制
2、基于EGFR小分子抑制剂
目前已经开发了四代EGFR小分子抑制剂。其中,已有三代针对EGFR的小分子抑制剂获批上市。根据结合口袋类型,EGFR抑制剂可分为靶向催化位点抑制剂和靶向变构位点抑制剂,前三代抑制剂都是靶向催化位点,第四代是靶向变构位点。
第一代EGFR抑制剂是基于嘧啶母核的吉非替尼和厄洛替尼,它们通过可逆地竞争TK催化区域中的Mg-ATP结合位点并随后阻断其信号转导。尽管会出现瘙痒和痤疮等剂量限制性毒性,但它们已被批准用于治疗EGFR L858R突变和EGFRdel19的NSCLC患者。
第二代抑制剂属于不可逆共价抑制剂,包括阿法替尼/达克替尼,结构中采用了迈克尔受体丙烯酰胺为共价基团,通过与TK活性位点的半胱氨酸形成共价键,增强了TK结构域和抑制剂之间的相互作用,解决了EGFRT790 M突变产生的耐药问题,但也存在选择性差等问题。
第三代EGFR抑制剂包括奥希替尼/奥莫替尼,奥希替尼也是一种不可逆的共价EGFR抑制剂,可以高效选择性地抑制EGFR突变体(外显子19缺失型EGFR、EGFR L858R/T790M ),但对EGFRWT的抑制作用较低。
不同于前三代小分子抑制剂,第四代抑制剂聚焦于靶向变构位点。其中,第四代EGFR抑制剂EAI045,是一种非ATP竞争性和突变选择性的变构抑制剂。研究发现,EAI045在单一疗法中无效,可能是因为EGFR被激活后二聚化使小分子抑制剂无法进入变构结合位点,从而限制了进一步的临床应用。JBJ-04-125-02是在EAI045基础上开发的具有更强抑制活性的小分子变构抑制剂。临床前研究表明,JBJ-04-125-02单药显示较好的肿瘤抑制效果。尽管四代EGFR抑制剂取得较大进展,但TKI的耐药问题不可避免地发生,仍需要努力寻找不同的抑制策略,以获得更有效的抑制剂来延缓甚至解决耐药性的发生。
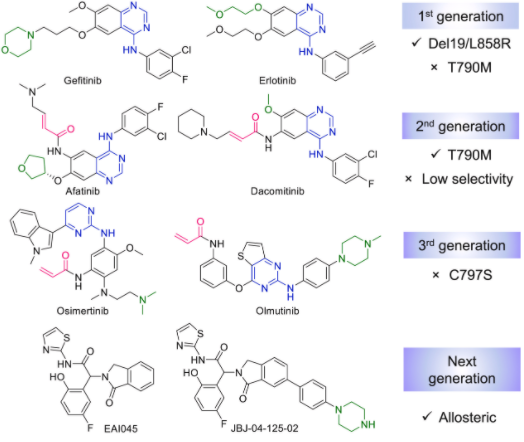
图2. 四代EGFR抑制剂的结构及面临问题
3、靶向EGFR的PROTAC研究进展
在过去数十年间,研究人员一共开发四代EGFR抑制剂。尽管取得了巨大的治疗成功,但这些EGFR 抑制剂的临床使用不可避免地导致获得性耐药,这对肿瘤靶向治疗提出了新的挑战。相较于小分子抑制剂来说,PROTAC是一种具有新结构新机制的肿瘤靶向药物,主要以事件驱动的作用方式来发挥作用,可以低剂量发挥较好疗效,避免了剂量依赖毒性的产生。PROTAC主要通过对致病靶蛋白进行多聚泛素化,进而诱导整个靶蛋白被泛素蛋白酶体系统降解,从而发挥治疗作用。对于克服耐药性以及不可成药靶点的药物研发具有重要优势。这对于易出现耐药的EGFR抑制剂来说,无疑是一种新型有效的研发策略。
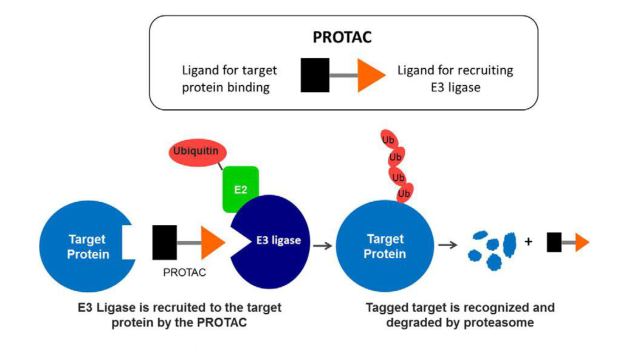
图3.PROTAC的作用机制
(1)、基于CRBN配体的EGFR PROTAC
2020年,西安交通大学张三奇课题组基于第四代EGFR-TKI 1与CRBN配体结合,设计合成了新型EGFR PROTAC(PROTAC 2),研究发现,PROTAC 2在HCC827细胞中可以有效诱导EGFR降解,DC50= 45.2 nM。并且后续的流式实验发现PROTAC 2可显着诱导HCC827细胞凋亡,使细胞停滞在G1期。
2020年,西奈山伊坎医学院金坚课题组以吉非替尼为靶头化合物,以沙利度胺为E3配体设计合成了EGFR PROTAC MS154。Western印迹分析实验表明,MS154可以有效诱导肺癌细胞中突变型 EGFR降解,并且比先前报道的EGFR降解剂更有效。此外,与母体药物吉非替尼相比,MS154抑制肿瘤细胞增殖活性优于母体化合物。
浙江工业大学张兴贤课题组开发了基于奥希替尼和来那度胺的新型EGFR降解剂16c,这是一种基于共价靶头化合物构建的PROTAC降解剂。并且化合物16c对PC9细胞、HCC827细胞和H1975细胞都显示出显著的抑制作用。Western印迹分析实验表明化合物16c可以通过泛素化有效降解EGFR蛋白,并在PC9细胞中达到最大降解率(Dmax= 68%)。同时,化合物16c可显著诱导PC9细胞凋亡,使细胞停滞在G0/G1期。
西安交通大学张三奇课题组报道了一种EGFR PROTAC(P3),研究发现,化合物P3可以有效诱导HCC827细胞中EGFR的降解,与AZD9291和靶头化合物相比,化合物P3 对HCC827细胞的增殖抑制作用更强。此外,化合物P3也可以有效降解突变型EGFRDel19和 EGFRL858R/T790M。
2021年,上海科技大学姜标课题组发现两种EGFR降解剂(PROTAC 3 和 PROTAC 4) ,主要是通过卡奈替尼和泊马度胺连接而成。研究发现,PROTAC 3 和 PROTAC 4在EGFR-TKI耐药肺癌细胞系中表现出强效和选择性的抗肿瘤活性,并可以选择性地降解H1975细胞突变型EGFRL858R/T790M和PC9细胞中的外显子19缺失型EGFREx19del。此外,与卡奈替尼相比,这些降解剂在H1975细胞和PC9Brca1 细胞中表现出更好的EGFR磷酸化抑制作用。

图4. 基于CRBN配体的EGFR PROTAC化学结构
上海科技大学姜标课题组报道了一种以布加替尼为靶头的EGFR PROTAC SIAIS164018。SIAIS164018能够有效降解突变的EGFRL858R + T790M和ALK融合蛋白(非小细胞肺癌中的两个最重要的靶标),并具有破坏转移相关癌蛋白的作用。在表达EGFR的H1975的细胞系和过表达ALKG1202R的293T细胞系中,SIAIS164018表现出比布加替尼更好的细胞增殖抑制作用。此外,SIAIS164018具有较好的口服生物利用度和良好的体内耐受性。
研究表明,缺氧是肿瘤细胞的共同特征。2021年郑州大学张晓坚课题组报道了一种缺氧激活的EGFR PROTAC(ha-PROTAC),通过将缺氧激活的离去基团4-nitrobenzyl 嵌入PROTAC,构建了一种内源性激活的前药PROTAC。蛋白降解实验发现,ha-PROTAC在低氧条件下对HCC4006细胞中的EGFRDel19表现出比在常氧条件下更强的降解活性。值得注意的是,这是利用肿瘤缺氧鉴定选择性作用于肿瘤的PROTACs的第一个例子,为PROTACs的开发提供了新的途径。
(2)、基于VHL配体的PROTAC
2018年,耶鲁大学Crews课题组报道了一种有效的EGFR降解剂PROTAC 5,由 EGFR激酶抑制剂拉帕替尼和VHL配体组成。PROTAC 5可以在OVCAR8细胞系中有效诱导降解EGFR,DC50=39.2 nM,Dmax= 97.6%。与拉帕替尼相比,具有更好的抗增殖作用。此外,PROTAC 5在SKBr3细胞中具有有效的抗增殖功效,IC50= 102 nM。重要的是,PROTAC 5 还可以诱导HeLa细胞系中外显子 20 插入突变型EGFR的降解。作者进一步采用吉非替尼作为靶头化合物开发PROTAC 6,可以降解HCC827细胞系中的EGFREx19del (DC50= 11.7 nM, Dmax= 98.9%) 和H3255细胞中的EGFRL858R (DC50= 22.3 nM和Dmax= 96.6%)。最后,作者设计了基于阿法替尼的PROTAC 7,PROTAC 7可以在H1975细胞系中降解吉非替尼耐药的突变型EGFRL858R/T790M,其DC50=215.8 nM和Dmax = 79.1%。
2020年,西安交通大学张三奇课题组开发了一种新型 EGFR PROTAC 10,化合物10在HCC827 细胞中诱导EGFRDel19的有效降解,DC50值为34.8 nM,Dmax=98%。此外,化合物10还可以显着诱导HCC827细胞的凋亡并将细胞停滞在G1期。
同年,伊坎医学院金坚课题组开发了基于吉非替尼的EGFR PROTAC MS39。蛋白质组学分析实验和蛋白降解实验表明,MS39显示出更好的靶点选择性和有效的蛋白降解效果,并有效抑制肺癌细胞的生长。此外,MS39在小鼠药代动力学研究中表现出较好生物利用度。值得注意的是,MS39是第一个适用于体内疗效研究的 EGFR PROTAC。
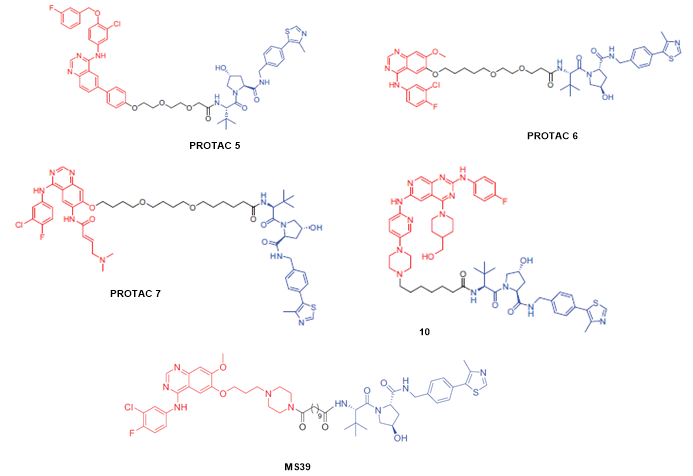
图5. 基于VHL配体的EGFR的化学结构
(3)、基于 IAPs的 PROTAC
2020年,南京医科大学周宏平课题组基于EGFRL858R/T790M选择性抑制剂XTF-262,设计合成了EGFRL858R/T790M突变的PROTACs/SNIPERs 14b。研究发现,基于VHL的 PROTAC能有效且选择性地降解EGFRL858R/T790M,DC50值为5.9 nM,而基于IAP的PROTAC 14b无法降解 EGFRL858R/T790M蛋白,具体原因还需进一步研究解决。
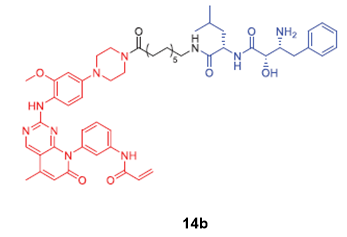
图6. 基于IAPs的EGFR PROTAC化学结构
(4)、靶向EGFR/PARP双靶点PROTAC 降解剂
华中科技大学李华课题组基于EGFR抑制剂吉非替尼和PARP抑制剂奥拉帕利为靶头化合物,合成不同链接长度和不同E3连接酶(CRBN-和VHL-)招募的EGFR和PARP蛋白双重靶向降解嵌合分子(Dual PROTACs),在细胞水平成功地同时降解癌细胞内的EGFR和PARP蛋白。
值得注意的是,这是Dual PROTACs第一个成功的例子,极大地拓展了PROTAC技术的应用,为PROTAC领域的研究提供了新思路。当然,Dual PROTACs存在分子量增大而导致透膜性差和药代动力学性质不佳等问题。要解决这些问题可能有两个策略,一个方向是采用纳米载药系统进行递送以改善药物的吸收,另一个方向就是精简抑制剂的结构,以缩小药物分子量。
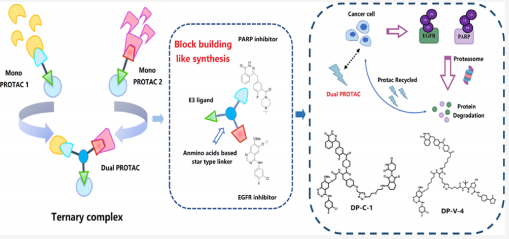
图7. PARP/EGFR双靶点PROTAC降解剂设计思路
— 小结 —
在过去数十年间,基于肺癌领域的药物研发取得了突破性成果,已经开发多种针对不同的EGFR突变类型EGFR抑制剂,用于治疗肺癌等疾病。正是由于这些药物的出现,极大地缓解了肺癌患者的痛苦,提高了患者的生活质量。但EGFR是一个极易发生突变的药物靶点,这些药物在临床使用中不可避免出现获得性耐药等问题。因此,迫切需要开发一种新机制的靶向EGFR的治疗策略。PROTAC是一种采用事件驱动的方式而不是占用驱动的药物类型。与传统小分子抑制剂相比,EGFR降解剂表现出良好的生物学结果,有可能解决耐药和EGFR代偿性上调等问题。目前,已经开发了多种EGFR的PROTAC降解剂,并且在临床前研究中表现出较好的治疗效果。
但基于EGFR的PROTAC研发同样面临巨大挑战。
1、有限可用的E3泛素连接酶配体。
2、PROTAC分子量大,可能会导致较差的透膜性及较差的ADMET性质。
3、缺乏完整系统的PROTAC设计技术。
4、PROTAC造成的毒性等不良反应。
目前基于EGFR PROTAC仍处于起步阶段,但靶向EGFR的PROTAC技术极具临床潜力,可能为显著提高EGFR抑制剂的选择性、疗效和耐药性提供新的治疗机会,为肺癌患者带来福音。
参考文献:
1.Rational Design and Synthesis of Novel Dual PROTACs for Simultaneous Degradation of EGFR and PARP. doi.org/10.1021/acs.jmedchem.1c00649. J. Med. Chem.2021, 64, 11, 7839–7852
2.Understanding and targeting resistance mechanisms in NSCLC.doi:10.1038/nrc.2017.84
3.Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol 2017;13:1667–21.
4.Small-molecule PROTACs: an emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 2018;36:553–62.
5.Specific non-genetic IAP-based protein erasers (SNIPERs) as a potential therapeutic strategy. Eur J Med Chem 2021;216:113247.
6.Recent advances in the development of EGFR degraders: PROTACs and LYTACs. doi.org/10.1016/j.ejmech.2022.114533
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言